
FunGen-xQTL Computational Protocol#
Developed for reproducible & reusable molecular QTL analyses for the NIH/NIA Alzheimer’s Disease Sequencing Project (ADSP) Functional Genomics xQTL (FunGen-xQTL) Project.
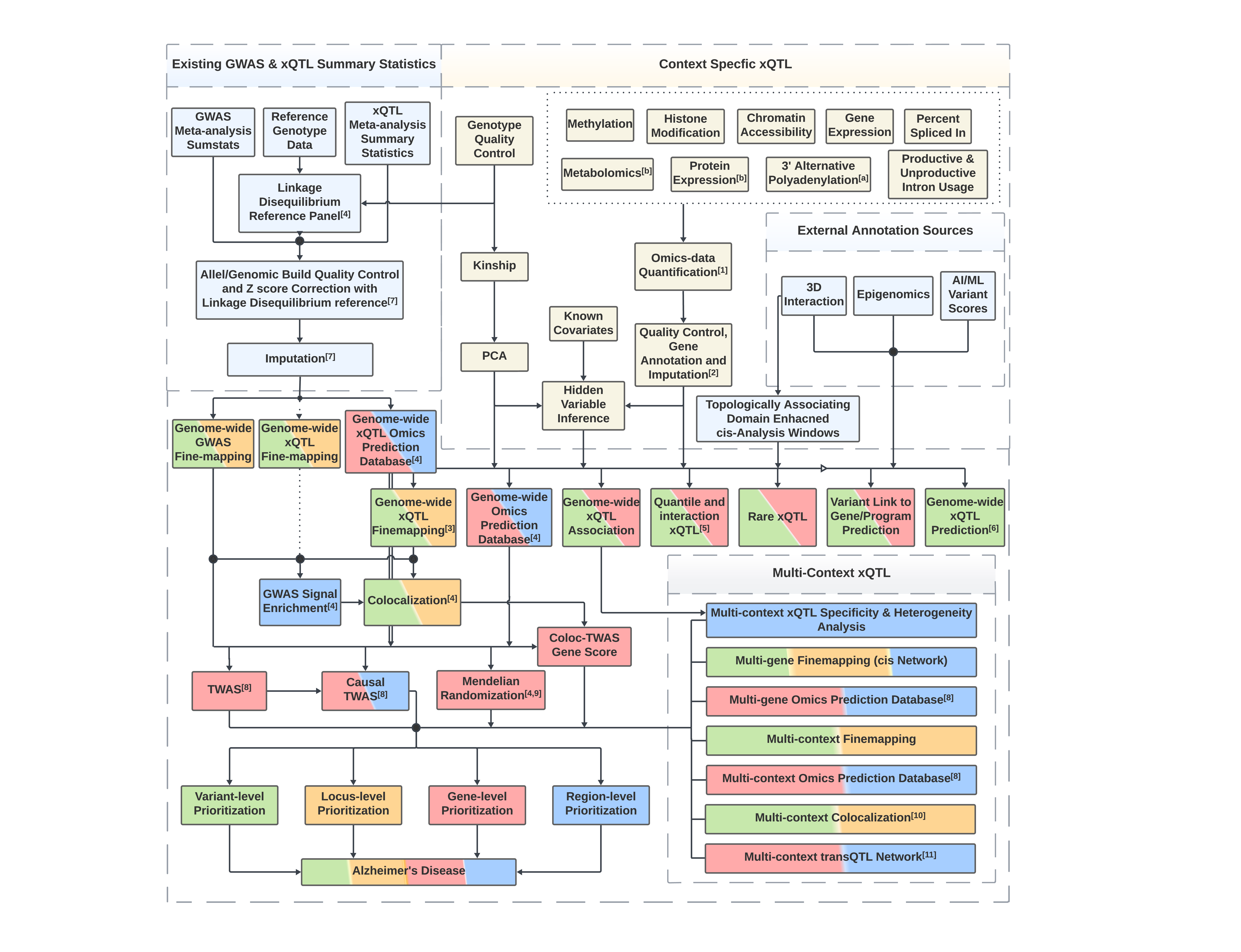
Overview of the protocol#
Standardized reference data#
Reference data are standardized and curated by the ADSP FGC Standardization Workgroup in coordination with NIAGCADS. Please find reference data specifications on ADSP Dashboard.
Software environment#
We have prepared containerized software environment through both Docker and Singularity virtualization systems to facilicate software environment setup and to aid in software reproducibility. For those not familiar with this concept please check out this wiki page of virtualization and an explanation on Docker website.
Pipeline execution#
Pipelines in this repository are written in the Script of Scripts (SoS) workflow language. Like most other workflow languages, SoS workflows can distribute and execute computing jobs directly in High Performance Computing cluster. It can also use containers (Docker or Singularity) to help with setting up computational environment and improve reproducibility. Unlike most other workflow languages, SoS workflows are created using SoS Notebooks (based on Ipython Notebook and developed in Jupyter) which allow for both scientific narrative and pipeline scripts in the same document. Unlike typical Jupyter Notebooks intended for interactive data analysis, SoS workflows written in Jupyter Notebooks can be executed directly as command line scripts either on a local computer or in a HPC environment.
We provide this toy example for running SoS pipeline on a typical HPC cluster environment. First time users are encouraged to try it out in order to help setting up the computational environment necessary to run the analysis in this protocol.
Source code#
Source code of pipelines and containers implemented in this repository are available at cumc/xqtl-protocol.
Container specifications for required software environments are available at cumc/xqtl-protocol.
How to use the resource#
Organization of the resource#
The website https://cumc.github.io/xqtl-protocol is generated from files under the code folder of the source code repository. The pipeline folder contains symbolic links automatically generated for pipeline files under code. The logic of the entire xQTL analysis workflow is roughly reflected on the left sidebar:
The GETTING STARTED section serves as the main landing page or index of the xQTL protocol, guiding users through the various pipelines implemented in this repository. It’s structured to mirror the logic of the xQTL analysis we’ve crafted. Because this page provides pointers to other sections, users can primarily focus here without having to sift through the rest of the pages pages in this repository.
The COMMAND GENERATOR section is designed as a one-stop hub for “push button” commands, enabling users to generate the full QTL analysis pipeline workflow scripts from a straightforward configuration file. Notebooks within this section are intended to be executed as command line software for data analysis command generation. Users can then execute these generated commands directly to conduct all preset analyses. Alternatively, they can tweak the commands to cater to particular analysis requirements. The configuration file serves a dual purpose: streamlining control and maintaining a record of executed workflows.
Other sections in bold fonts provide an array of available analyses, presented roughly from upstream to downstream processes. Most of these sections feature mini-protocols, represented as clickable, non-bold text under each analysis category, leading to specific notebooks. These notebooks detail the commands necessary for the analyses defined in the respective mini-protocols. Predominantly tutorial-based, they are designed to be executed interactively in Jupyter or via the command terminal, allowing users to navigate through the SoS pipelines step by step. A few of these sections serve as actual pipeline modules which we’ll discuss next (see below).
Mini-protocols, as mentioned earlier, can be expanded by clicking the downward arrows, revealing the SoS implementations of pipeline modules. These represent the crux of the pipeline implementations and are intended to be executed as command line software. They’re also self-contained, allowing for reusability beyond the specific context of xQTL data analysis.
Computing environment setup#
In order to run the xQTL protocol on your computer (or a High Performance Computing cluster), please install Script of Scripts (version >= 0.24.1) (see here for a tutorial to set it up with
micromamba).For Linux and Mac desktop users you can either install the container
SingularityorDocker. In the xQTL project we useSingularity. Here are some tips to set up Singularity on MacOS.For Linux-based HPC users, your system may already have
Singularityinstalled. If not please communicate with the IT support for the HPC. Typically Docker is not allowed on HPC.For Windows users, you will need to install WSL (we have tested it on WSL2 and not on WSL1) and then install
Singularitywithin WSL as instructed in this post.
We have provided example data-sets and
Singularitycontainer images in this Synapse folder. For guidance on downloading the data programmatically, refer to this documentation. If you need to set up a Synapse client, consult this guide.Within the
test_datafolder, datasets prefixed with MWE (Minimal Working Example) are provided. These are used for unit testing each module, ensuring the integrity of the code.
The
protocol_datafolder houses a comprehensive set of data, illustrating the full extent of our protocol. This is showcased in this notebook, with the source code available for reference.The
container/singularityfolder contains the released Singularity images for the software environment. For Docker users (e.g., on Linux or Mac Desktops), downloading this folder is not necessary.Please clone this repository cumc/xqtl-protocol onto your computer. This is the source code for this resource. All pipelines are symbolic links in the
pipelinefolder. Users are encouraged to execute from the root of the repository folders by typing
sos run pipeline/<pipeline_file>.ipynb
that is, executing the symbolic links directly to perform the analysis.
See Also#
Analysis from FunGen-xQTL consortium using this protocol can be found at cumc/fungen-xqtl-analysis
Our team#
This repository is developed by the Analysis Working Group of the NIA FunGen-xQTL consortium.
Developers#
Lead developers
Hao Sun, Department of Neurology, Columbia University
Gao Wang, Department of Neurology, Columbia University
Main contributors (largely based on GitHub Pull Requests)
Name |
Affiliation |
|---|---|
Xuanhe Chen |
Department of Biostatistics, Columbia University |
Wenhao Gou |
Department of Biostatistics, Columbia University |
Liucheng Shi |
Department of Biostatistics, Columbia University |
Haochen Sun |
Department of Biostatistics, Columbia University |
Zining Qi |
Department of Biostatistics, Columbia University |
Yifei Wang |
Department of Neurology, Columbia University |
Ru Feng |
Department of Neurology, Columbia University |
Alexandre Pelletier |
Department of Medicine, Boston University |
Travyse Edwards |
Mount Sinai & University of Pennsylvania |
Daniel Nachun |
Department of Pathology, Stanford University |
Jiacheng Li |
Department of Neurology, Columbia University |
Mintao Lin |
Department of Medicine, Boston University |
Leadership#
FunGen-AD
Name |
Affiliation |
|---|---|
Philip De Jager |
Department of Neurology, Columbia University |
Carlos Crunchaga |
Department of Psychiatry, Neurology and Genetics, Washington University in St. Louis |
FunGen-xQTL Analysis Working Group
Name |
Affiliation |
|---|---|
Gao Wang |
Department of Neurology, Columbia University |
Xiaoling Zhang |
Departments of Medicine and Biostatistics, Boston University |
Edoardo Marcora |
Departments of Neuroscience, Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai |
Fanny Leung |
Department of the Pathology and Laboratory Medicine, University of Pennsylvania |
Julia TCW |
Department of Pharmacology and Bioinformatics, Boston University |
Kushal K. Dey |
Memorial Sloan Kettering |
Alan Renton |
Departments of Neuroscience, Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai |
Stephen Montgomery |
Department of Pathology, Stanford University |
Xiaoquan Wen |
Department of Biostatistics, University of Michigan |