
APA Calling#
Aim#
The purpose of this notebook is to call APA-based information (PDUI) based on DAPARS2 method.
Methods#
%preview ../../images/apa_calling.png
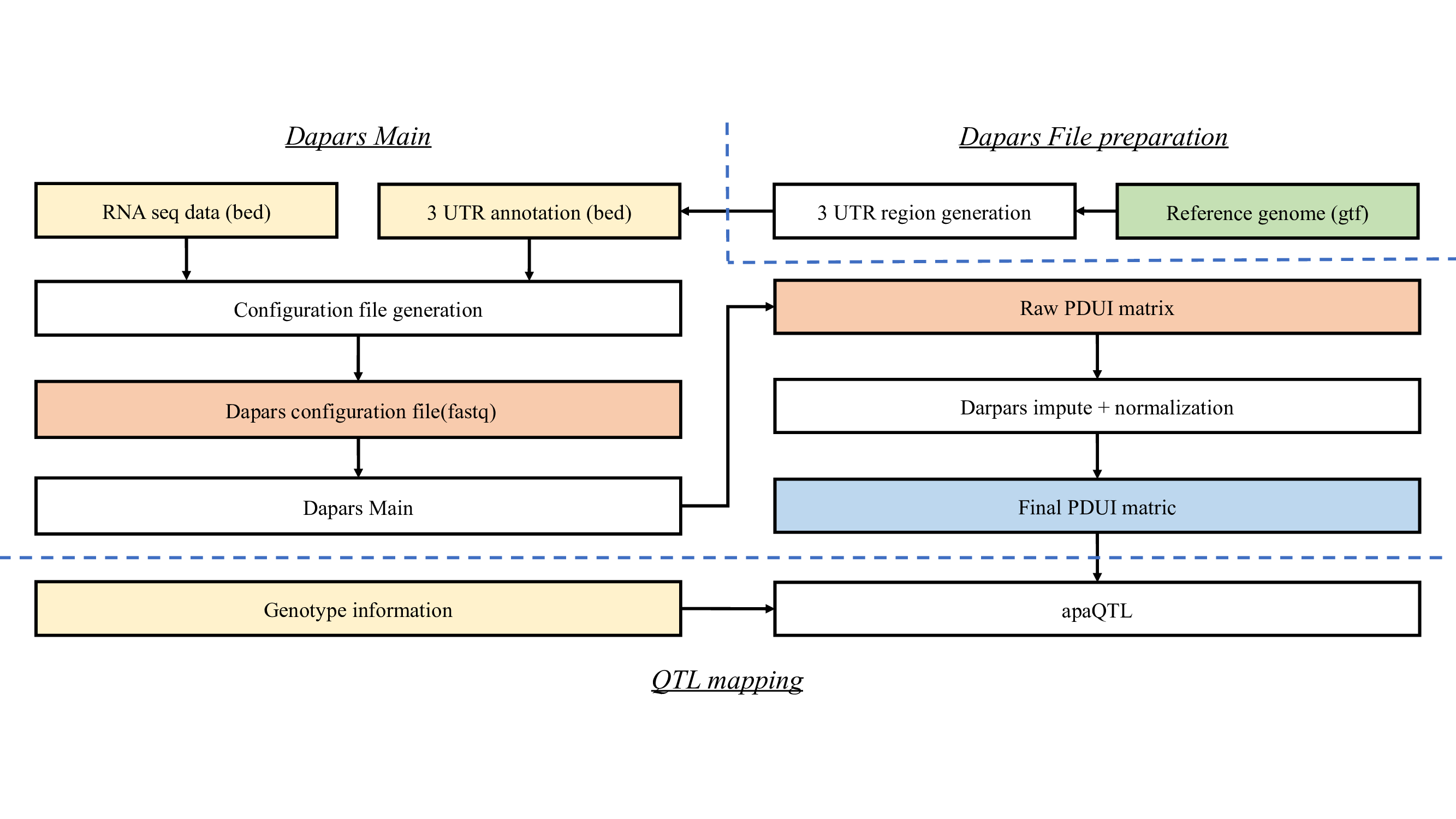
3’UTR Reference#
gtf2bed12.py : Covert gtf to bed format (Source from in-house codes from Li Lab: Xu-Dong/Exon_Intron_Extractor)
DaPars_Extract_Anno.py : extract the 3UTR regions in bed formats from the whole genome bed (Source from Dapars 2: 3UTR/DaPars2)
Call WIG data from transcriptome BAM files#
Using bedtools or rsem-bam2wig, for RSEM based alignment
Config files Generation#
Python 3 loops to read line by line the sum of reads coverage of all chromosome.
Dapars2 Main Function#
Dapars2_Multi_Sample.py: use the least sqaures methods to calculate the usage of long isoforms (3UTR/DaPars2)
Note: this part of code have been modified from source to deal with some formatting discrepancy in wig file
Impute missing values in Dapars result#
KNN using impute R package.
Input#
A list of transcriptome level BAM files, eg generated by RSEM
The 3’UTR annotation reference file
If you do not have 3’UTR annotation file, please generate it first. Input to this step is the transcriptome level gene feature file in GTF format that we previously prepared.
Output#
Dapars config files
PUDI (Raw) information saved in txt
PDUI (Imputed) information saved in txt. This is recommended for further analysis.
Minimal working example#
To generate 3’UTR reference data,
sos run apa_calling.ipynb UTR_reference \
--cwd output/apa \
--hg-gtf reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.ERCC.gtf \
--container /mnt/mfs/statgen/ls3751/container/dapars2_final.sif
Command interface#
sos run apa_calling.ipynb -h
usage: sos run apa_calling.ipynb [workflow_name | -t targets] [options] [workflow_options]
workflow_name: Single or combined workflows defined in this script
targets: One or more targets to generate
options: Single-hyphen sos parameters (see "sos run -h" for details)
workflow_options: Double-hyphen workflow-specific parameters
Workflows:
UTR_reference
bam2tools
bam2toolsv1
bam2toolsv2
bam2toolstmp
APAconfig
APAmain
Global Workflow Options:
--walltime 400h
--mem 200G
--ncore 16 (as int)
--cwd output (as path)
the output directory for generated files
--numThreads 8 (as int)
Number of threads
--job-size 1 (as int)
--container ''
Sections
UTR_reference: Generate the 3UTR region according to the gtf file
Workflow Options:
--hg-gtf VAL (as path, required)
gtf file
bam2tools:
Workflow Options:
--n 9 (as int)
bam2toolsv1:
Workflow Options:
--sample VAL (as path, required)
--tissue VAL (as path, required)
bam2toolsv2:
Workflow Options:
--sample VAL (as path, required)
--tissue VAL (as path, required)
bam2toolstmp:
Workflow Options:
--sample VAL (as path, required)
--tissue VAL (as path, required)
APAconfig: Generate configuration file
Workflow Options:
--bfile VAL (as path, required)
--annotation VAL (as path, required)
--least-pass-coverage-percentage 0.3 (as float)
Default parameters for Dapars2:
--coverage-threshold 10 (as int)
APAmain: Call Dapars2 multi_chromosome
Workflow Options:
--[no-]chr-prefix (default to False)
--chrlist VAL VAL ... (as type, required)
Workflow implementation#
[global]
parameter: walltime = '400h'
parameter: mem = '200G'
parameter: ncore = 16
# the output directory for generated files
parameter: cwd = path("output")
# Number of threads
parameter: numThreads = 8
parameter: job_size = 1
parameter: container = ''
import re
parameter: entrypoint= ('micromamba run -a "" -n' + ' ' + re.sub(r'(_apptainer:latest|_docker:latest|\.sif)$', '', container.split('/')[-1])) if container else ""
Step 0: Generate 3UTR regions based on GTF#
The 3UTR regions (saved in bed format) could be use repeatly for different samples. It only served as the reference region. You may not need to run it if given generated hg19/hg38 3UTR regions.
# Generate the 3UTR region according to the gtf file
[UTR_reference]
# gtf file
parameter: hg_gtf = path
input: hg_gtf
output: f'{cwd}/{_input:bn}.bed',
f'{cwd}/{_input:bn}.transcript_to_geneName.txt',
f'{cwd}/{_input:bn}_3UTR.bed'
bash: expand = '${ }', container = container
gtf2bed12.py --gtf ${_input} --out ${cwd}
mv ${cwd}/gene_annotation.bed ${_output[0]}
mv ${cwd}/transcript_to_geneName.txt ${_output[1]}
DaPars_Extract_Anno.py -b ${_output[0]} -s ${_output[1]} -o ${_output[2]}
Step 1: Generate WIG calls and flagstat files from BAM files#
Generating WIG from BAM data via bedtools is recommended by Dapars authors. However, our transcriptome level calls are made using RSEM, which in fact contains a program called rsem-bam2wig with this one additional feature:
–no-fractional-weight : If this is set, RSEM will not look for “ZW” tag and each alignment appeared in the BAM file has weight 1. Set this if your BAM file is not generated by RSEM. Please note that this option must be at the end of the command line
Here we stick to bedtools because of its popularity and most generic BAM files may not have the ZW tag anyways.
[bam2tools]
parameter: n = 9
n = [x for x in range(n)]
input: for_each = 'n'
task: trunk_workers = 1, trunk_size = job_size, walltime = walltime, mem = mem, cores = numThreads
python: expand = True, container = container
import glob
import os
import subprocess
path = "/mnt/mfs/ctcn/datasets/rosmap/rnaseq/dlpfcTissue/batch{_n}/STAR_aligned"
name = glob.glob(path + "/**/*Aligned.sortedByCoord.out.bam", recursive = True)
wigpath = "/home/ls3751/project/ls3751/wig/batch{_n}/"
if not os.path.exists(wigpath):
os.makedirs(os.path.dirname(wigpath))
for i in name:
id = i.split("/")[-2]
filedir = path + "/" + id + "/" + id + ".bam"
out = wigpath + id + ".wig"
new_cmd = "bedtools genomecov -ibam " + filedir + " -bga -split -trackline" + " > " + out
os.system(new_cmd)
out_2 = wigpath + id + ".flagstat"
new_cmd_2 = f"samtools flagstat --thread {numThreads} " + filedir + " > " + out_2
os.system(new_cmd_2)
[bam2toolsv1]
parameter: sample = path
parameter: tissue = path
task: trunk_workers = 1, trunk_size = job_size, walltime = walltime, mem = mem, cores = numThreads, concurrent = True
python: expand = "${ }", container = container
import os
import multiprocessing
import re
sample_list = [line.strip('\n') for line in open("${sample}")]
def call_wig_flagstat(subject):
id = re.findall(r"\D(\d{8})\D",subject)[1]
prefix = "/mnt/mfs/statgen/ls3751/aqtl_analysis/wig/" + "${tissue}" + "/"
out = prefix + id + ".wig"
out_2 = prefix + id + ".flagstat"
new_cmd = "bedtools genomecov -ibam " + subject + " -bga -split -trackline" + " > " + out
os.system(new_cmd)
new_cmd_2 = f"samtools flagstat --thread {numThreads} " + subject + " > " + out_2
os.system(new_cmd_2)
for sample in sample_list:
call_wig_flagstat(sample)
### process = multiprocessing.Process(target = call_wig_flagstat, args =(sample_list[i]))
### process.start()
### for p in processes:
### p.join()
[bam2toolsv2]
parameter: sample = path
parameter: tissue = path
task: trunk_workers = 1, trunk_size = job_size, walltime = walltime, mem = mem, cores = numThreads
bash: expand = "${ }", container = container
input="${sample}"
while IFS=',' read -r col1 col2
do
bedtools genomecov -ibam $col1 -bga -split -trackline > /mnt/mfs/statgen/ls3751/aqtl_analysis/wig/${tissue}/$col2.wig &
samtools flagstat --thread ${numThreads} $col1 > /mnt/mfs/statgen/ls3751/aqtl_analysis/wig/${tissue}/$col2.flagstat &
done < "$input"
[bam2toolstmp]
parameter: sample = path
parameter: tissue = path
task: trunk_workers = 1, trunk_size = job_size, walltime = walltime, mem = mem, cores = ncore
python: expand = "${ }", container = container
import os
import re
sample_list = [line.strip('\n') for line in open("${sample}")]
def copy_wigfile(subject):
id = re.findall(r"\D(\d{8})\D",subject)[0]
prefix = "/mnt/mfs/statgen/ls3751/aqtl_analysis/wig/" + "${tissue}" + "/"
file1 = os.path.splitext(subject)[0] + ".flagstat"
out = prefix + id + ".wig"
out_2 = prefix + id + ".flagstat"
new_cmd = "cp " + subject + " " + out
os.system(new_cmd)
new_cmd_2 = "cp " + file1 + " " + out_2
os.system(new_cmd_2)
for sample in sample_list:
copy_wigfile(sample)
sos run /mnt/mfs/statgen/ls3751/github/xqtl-protocol/code/molecular_phenotypes/calling/apa_calling.ipynb bam2toolsv2 --cwd /mnt/mfs/statgen/ls3751/aqtl_analysis/wig/scripts --sample /mnt/mfs/statgen/ls3751/aqtl_analysis/wig/scripts/AC_1.txt --tissue AC --container /mnt/mfs/statgen/ls3751/container/dapars2_final.sif -c /home/ls3751/project/ls3751/csgg.yml
INFO: Running bam2toolsv2:
INFO: bam2toolsv2 is completed.
INFO: Workflow bam2toolsv2 (ID=wdde1442ae33498bb) is executed successfully with 1 completed step.
Step 2: Generating config files and calculating sample depth#
Notes on input file format#
For the input file, it has the following format. Additional notes are:
The first line is the information of file. If you do not have them, please add any content on first line
The file must end with “.wig”. It will not cause any problem if you directly change from “.bedgraph”
If your input wig file did not have the characters “chr” in the first column, please set
no_chr_prefix = T
head -n 10 /mnt/mfs/statgen/ls3751/MWE_dapars2/sample1.wig
track type=bedGraph
1 0 10099 0
1 10099 10113 1
1 10113 10146 0
1 10146 10157 2
1 10157 10230 0
1 10230 10241 2
1 10241 10279 0
1 10279 10301 1
1 10301 10329 0
# Generate configuration file
[APAconfig]
parameter: bfile = path
parameter: annotation = path
parameter: job_size = 1
# Default parameters for Dapars2:
parameter: least_pass_coverage_percentage = 0.3
parameter: coverage_threshold = 10
output: [f'{cwd}/sample_mapping_files.txt',f'{cwd}/sample_configuration_file.txt']
task: trunk_workers = 1, trunk_size = 1, walltime = walltime, mem = mem, cores = ncore
python3: expand = "${ }", container = container
import re
import os
target_all_sample = os.listdir("${bfile}")
target_all_sample = list(filter(lambda v: re.match('.*wig$', v), target_all_sample))
target_all_sample = ["${bfile}" + "/" + w for w in target_all_sample]
def extract_total_reads(input_flagstat_file):
num_line = 0
total_reads = '-1'
#print input_flagstat_file
for line in open(input_flagstat_file,'r'):
num_line += 1
if num_line == 5:
total_reads = line.strip().split(' ')[0]
break
return total_reads
#print(target_all_sample)
print("INFO: Total",len(target_all_sample),"samples found in provided dirctory!")
# Total depth file:
mapping_file = open("${_output[0]}", "w")
for current_sample in target_all_sample:
flag = current_sample.split(".")[0] + ".flagstat"
current_sample_total_depth = extract_total_reads(flag)
field_out = [current_sample, str(current_sample_total_depth)]
mapping_file.writelines('\t'.join(field_out) + '\n')
print("Coverage of sample ", current_sample, ": ", current_sample_total_depth)
mapping_file.close()
# Configuration file:
config_file = open(${_output[1]:r},"w")
config_file.writelines(f"Annotated_3UTR=${annotation}\n")
config_file.writelines( "Aligned_Wig_files=%s\n" % ",".join(target_all_sample))
config_file.writelines(f"Output_directory=${cwd}/apa \n")
config_file.writelines(f"Output_result_file=Dapars_result\n")
config_file.writelines(f"Least_pass_coverage_percentage=${least_pass_coverage_percentage}\n")
config_file.writelines( "Coverage_threshold=${coverage_threshold}\n")
config_file.writelines( "Num_Threads=${numThreads}\n")
config_file.writelines(f"sequencing_depth_file=${_output[0]}")
config_file.close()
Step 3: Run Dapars2 main to calculate PDUIs#
Default input of Dapars2_Multi_Sample.py did not consider the situation that first column did not contain “chr” (shown in Step 2). We add a new argument no_chr_prefix (default is FALSE)
# Call Dapars2 multi_chromosome
[APAmain]
parameter: chr_prefix = False
parameter: chrlist = list
input: for_each = 'chrlist'
output: [f'{cwd}/apa_{x}/Dapars_result_result_temp.{x}.txt' for x in chrlist], group_by = 1
task: trunk_workers = 1, trunk_size = job_size, walltime = walltime, mem = mem, cores = ncore
bash: expand = True, container = container
python2 /mnt/mfs/statgen/ls3751/github/xqtl-protocol/code/Dapars2_Multi_Sample.py {cwd}/sample_configuration_file.txt {_chrlist} {"F" if chr_prefix else "T"}
Analysis demo#
Step 0: 3UTR generation#
sos run /mnt/mfs/statgen/ls3751/github/xqtl-protocol/code/molecular_phenotypes/calling/apa_calling.ipynb UTR_reference \
--cwd /mnt/mfs/statgen/ls3751/MWE_dapars2/Output \
--hg_gtf /mnt/mfs/statgen/ls3751/MWE_dapars2/gencode.v39.annotation.gtf \
--container /mnt/mfs/statgen/ls3751/container/dapars2_final.sif
INFO: Running UTR_reference: Generate the 3UTR region according to the gtf file
Done!
Generating regions ...
Total extracted 3' UTR: 185865
Finished
INFO: UTR_reference is completed.
INFO: UTR_reference output: /mnt/mfs/statgen/ls3751/MWE_dapars2/Output/gencode.v39.annotation.bed /mnt/mfs/statgen/ls3751/MWE_dapars2/Output/gencode.v39.annotation.transcript_to_geneName.txt... (3 items)
INFO: Workflow UTR_reference (ID=w907045e2667aefaa) is executed successfully with 1 completed step.
tree /mnt/mfs/statgen/ls3751/MWE_dapars2/Output
/mnt/mfs/statgen/ls3751/MWE_dapars2/Output
├── gencode.v19.annotation_3UTR.bed
├── gene_annotation.bed
└── transcript_to_geneName.txt
0 directories, 3 files
Step 1: Bam to wig process and extracting reads depth file#
sos run /mnt/mfs/statgen/ls3751/github/xqtl-protocol/code/molecular_phenotypes/calling/apa_calling.ipynb bam2tools \
--n 0 1 2 3 4 5 6 7 8 \
--container /mnt/mfs/statgen/ls3751/container/dapars2_final.sif
sos run /mnt/mfs/statgen/ls3751/github/xqtl-protocol/code/molecular_phenotypes/calling/apa_calling.ipynb bam2toolsv2 \
--cwd /mnt/mfs/statgen/ls3751/aqtl_analysis \
--sample /mnt/mfs/statgen/ls3751/aqtl_analysis/wig/scripts/AC_1.txt \
--tissue AC \
--container /mnt/mfs/statgen/ls3751/container/dapars2_final.sif
Step 2: Generating config files#
sos run /mnt/mfs/statgen/ls3751/github/xqtl-protocol/code/molecular_phenotypes/calling/apa_calling.ipynb APAconfig \
--cwd /mnt/mfs/statgen/ls3751/rosmap/dlpfcTissue/batch0 \
--bfile /mnt/mfs/statgen/ls3751/rosmap/dlpfcTissue/batch0 \
--annotation /mnt/mfs/statgen/ls3751/MWE_dapars2/Output/gencode.v39.annotation_3UTR.bed \
--container /mnt/mfs/statgen/ls3751/container/dapars2_final.sif
INFO: Running APAconfig: Calculcate total depth and configuration file
INFO: Total 4 samples found in provided dirctory!
Coverage of sample /mnt/mfs/statgen/ls3751/MWE_dapars2/sample4.wig : 5943757290
Coverage of sample /mnt/mfs/statgen/ls3751/MWE_dapars2/sample3.wig : 6060347021
Coverage of sample /mnt/mfs/statgen/ls3751/MWE_dapars2/sample2.wig : 3617065465
Coverage of sample /mnt/mfs/statgen/ls3751/MWE_dapars2/sample1.wig : 2573149742
INFO: APAconfig is completed.
INFO: APAconfig output: /mnt/mfs/statgen/ls3751/MWE_dapars2/Output/sample_mapping_files.txt /mnt/mfs/statgen/ls3751/MWE_dapars2/Output/sample_configuration_file.txt
INFO: Workflow APAconfig (ID=wdc3865265bf562ea) is executed successfully with 1 completed step.
tree /mnt/mfs/statgen/ls3751/MWE_dapars2/Output
/mnt/mfs/statgen/ls3751/MWE_dapars2/Output
├── gencode.v19.annotation_3UTR.bed
├── gene_annotation.bed
├── sample_configuration_file.txt
├── sample_mapping_files.txt
└── transcript_to_geneName.txt
0 directories, 5 files
Step 3: Dapars2 Main#
Note: the example is a truncated version, which just have coverage in chr1,chr11 and chr12
sos run /mnt/mfs/statgen/ls3751/github/xqtl-protocol/code/molecular_phenotypes/calling/apa_calling.ipynb APAmain \
--cwd /mnt/mfs/statgen/ls3751/rosmap/dlpfcTissue/batch0 \
--chrlist chr21 chr14 chr1 \
--container /mnt/mfs/statgen/ls3751/container/dapars2_final.sif
INFO: Running APAmain: Call Dapars2 multi_chromosome
[Sun Jan 30 04:15:34 2022] Start Analysis ...
All samples Joint Processing chr1 ...
[Sun Jan 30 04:15:34 2022] Loading Coverage ...
/mnt/mfs/statgen/ls3751/MWE_dapars2/sample4.wig
/mnt/mfs/statgen/ls3751/MWE_dapars2/sample3.wig
/mnt/mfs/statgen/ls3751/MWE_dapars2/sample2.wig
/mnt/mfs/statgen/ls3751/MWE_dapars2/sample1.wig
[5943757290 6060347021 3617065465 2573149742]
[Sun Jan 30 04:15:46 2022] Loading Coverage Finished ...
#assigned events:
1379
1379
1379
1379
1379
1378
1378
1378
[Sun Jan 30 04:15:51 2022] Finished!
INFO: APAmain is completed.
INFO: APAmain output: /mnt/mfs/statgen/ls3751/MWE_dapars2/Output/Wigfiles_chr1/Dapars_result_result_temp.chr1.txt
INFO: Workflow APAmain (ID=w28477dad0713d81d) is executed successfully with 1 completed step.
tree /mnt/mfs/statgen/ls3751/MWE_dapars2/Output
/mnt/mfs/statgen/ls3751/MWE_dapars2/Output
├── gencode.v19.annotation_3UTR.bed
├── gene_annotation.bed
├── sample_configuration_file.txt
├── sample_mapping_files.txt
├── transcript_to_geneName.txt
└── Wigfiles_chr1
├── Dapars_result_result_temp.chr1.txt
└── tmp
├── Each_processor_3UTR_Result_1.txt
├── Each_processor_3UTR_Result_2.txt
├── Each_processor_3UTR_Result_3.txt
├── Each_processor_3UTR_Result_4.txt
├── Each_processor_3UTR_Result_5.txt
├── Each_processor_3UTR_Result_6.txt
├── Each_processor_3UTR_Result_7.txt
└── Each_processor_3UTR_Result_8.txt
2 directories, 14 files